 云霞育儿网
云霞育儿网2 电子 versus 4 电子:深入解读二者的不同之处
两电子ORR通过优化ΔG(*OOH)控制H₂O₂选择性,依赖氮掺杂或单原子催化剂(如FeN₄)调控d带中心抑制四电子路径。
四电子ORR需平衡O*与OH*的自由能差(ΔG_O-ΔG_OH),通过合金化(Pt3Co)或应变工程提升效率。DFT计算火山图与d带中心理论指导催化剂设计,揭示中间体吸附能与反应路径关联。
显式溶剂模型修正界面效应,非贵金属催化剂(如Cr-O₂)突破标度关系,推动ORR从经验试错转向理性设计。
什么是两电子ORR?
两电子氧还原反应(2e⁻ORR)是氧气在催化剂表面通过两电子转移路径部分还原生成过氧化氢(H₂O₂)的关键过程,其反应路径的模拟与催化剂设计高度依赖密度泛函理论(DFT)计算。
DFT通过构建催化剂表面模型,分析氧气分子(O₂)的吸附构型、中间体稳定性和反应能垒,揭示2e⁻ORR的选择性来源。
具体而言,氧气分子首先吸附于催化剂表面形成O₂*,随后通过质子-电子协同转移生成关键中间体*OOH,其结合自由能(ΔG(*OOH))是决定反应路径的核心参数。
若ΔG(*OOH)过强,*OOH的O-O键易断裂,反应转向四电子路径生成H₂O;若ΔG(*OOH)适中,*OOH进一步还原为H₂O₂,完成2e⁻路径。
DFT计算通过构建火山图(Volcano Plot),将ΔG(*OOH)与催化活性关联,发现当ΔG(*OOH)接近0 eV时,催化剂的活性和选择性最优,此时中间体的吸附与脱附达到动态平衡。

DOI:10.1002/advs.202100076
在催化剂设计方面,DFT揭示了电子结构调控对2e⁻ORR的调控机制。
例如,碳基材料中氮掺杂或缺陷结构可调节活性位点的电荷分布,优化OOH的结合强度;金属单原子催化剂(如FeN₄)通过调整d带中心位置,削弱氧中间体的吸附,从而抑制四电子路径。
DFT还量化了合金化或双金属位点的协同效应,如PdAu合金表面通过电子再分配降低OOH生成能垒,显著提升H₂O₂选择性。
此外,DFT模拟溶剂化效应和电极-电解质界面结构,揭示了真实反应环境中中间体稳定性的动态变化,为修正理论模型提供了依据。

DOI:10.1039/d3sc00250k
尽管DFT在预测催化剂活性和路径选择上具有优势,其局限性在于对复杂界面环境的简化。例如,传统计算常忽略溶剂分子和电场对中间体稳定性的影响,导致预测与实验偏差。
未来需发展多尺度模型,结合机器学习加速筛选,以实现更精准的催化剂设计。
总之,DFT通过原子尺度解析反应机制,为2e⁻ORR的高效催化剂开发提供了理论基石,推动了从“试错法”向“理性设计”的转变。
什么是四电子ORR?
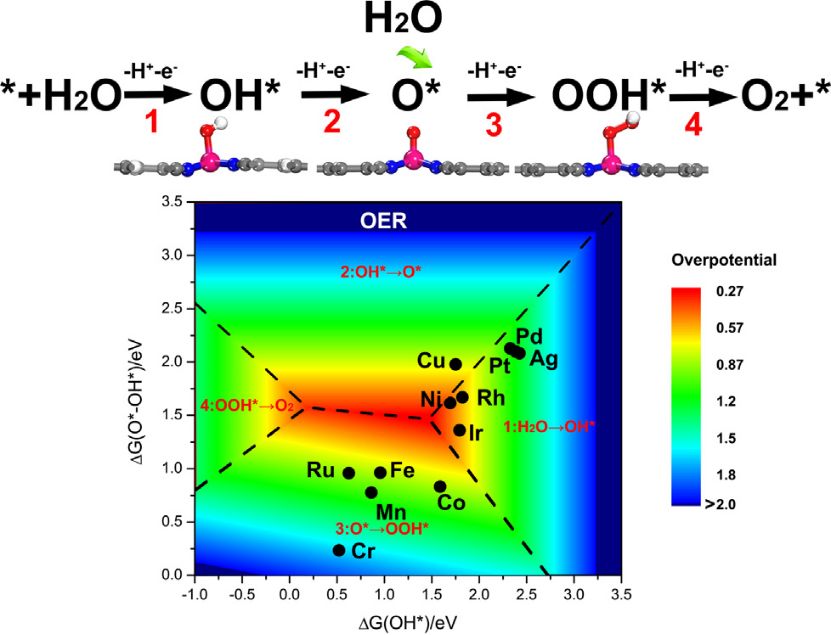
四电子氧还原反应(ORR)是燃料电池等能源装置中氧气分子通过四个电子转移路径高效转化为水的核心过程,其动力学效率直接受催化剂表面中间体吸附行为调控。基于密度泛函理论(DFT)的计算研究表明,四电子ORR遵循质子-电子耦合转移机制,涉及OOH、O和OH三个关键中间体:氧气分子首先吸附于催化剂表面形成O₂,随后通过质子与电子的逐步加入,依次转化为过氧中间体(OOH*)、原子氧中间体(O*)和羟基中间体(OH*),最终脱附生成水。
DFT通过构建催化剂表面模型,计算各步骤的吉布斯自由能(ΔG),揭示反应能垒与过电位(η)的关联性。
例如,在酸性条件下,理想催化剂的ΔG应满足各步骤自由能变化均接近1.23 eV,此时过电位趋近于零。
火山图分析显示,中间体O与OH的自由能差值(ΔG_O*-ΔG_OH*)是决定η的核心参数,其最优范围对应火山顶点,如Pt因O*吸附能适中而位于高效区。
DOI:10.1016/j.jcat.2018.12.012
DFT进一步通过d带中心理论揭示金属活性位点的电子结构对吸附强度的影响:过渡金属的d轨道与氧分子反键轨道杂化程度越高,中间体吸附越强。例如,Fe-N4单原子催化剂中,Fe的d带中心位置通过配位环境调控,使OH的吸附自由能(ΔG_OH)降至0.35 eV,突破传统标度关系限制。
此外,DFT模拟溶剂化效应与界面电场对质子转移速率的影响,发现缓慢的质子动力学更利于O-O键断裂,从而抑制双电子副反应。
催化剂设计中,DFT指导的合金化(如Pt3Co)与应变工程(如核壳结构Au@Pt)可优化d带中心位置,降低速率决定步骤(RDS)能垒。
对于非贵金属催化剂,计算表明氧配位的Cr-O2位点可通过锯齿边缘的电子再分布增强O₂吸附,其四电子路径选择性显著高于传统氮配位结构。这些理论成果为高效ORR催化剂的理性设计提供了原子尺度蓝图。

DOI:10.20517/energymater.2023.52

发表评论:
◎欢迎参与讨论,请在这里发表您的看法、交流您的观点。