Claude 是由Anthropic公司开发,基于自然语言处理的大型语言模型,拥有强大的理解和生成能力。最初仅限于文本生成,逐步发展至如今能够处理更复杂的任务,包括编程、数据分析等。
Claude的核心理念是通过自然语言和用户交互,使科研分析变得更直观、简单。现在,它已经能够执行复杂的生信分析任务,并能为科研人员提供实时反馈,自动生成分析报告。
🚀 Claude对生信领域的益处传统的生信分析流程往往涉及到大量的命令行操作和复杂的编程,门槛高,学习曲线陡峭。而Claude通过与用户对话,自动完成数据分析的所有步骤,极大地简化了分析过程。
Claude的优势包括:
零门槛:不需要学习复杂的命令和代码,直接通过自然语言和Claude对话完成分析。提高效率:自动化的分析流程让科研人员能够专注于数据解读和科学创新。降低错误率:自动生成的命令和分析步骤经过精心设计,减少人为错误的发生。❝💡现在已经率先部署Claude模型,并开通了API接口,方便大家更简单,更高效的完成自己的数据分析!
一个完整的示例教程你是否一直想做生信数据分析,却苦于不会 Linux、不会代码、不懂流程?今天小编来手把手带你用 Claude 完成全流程分析
👉你完全不需要敲命令
👉你只需要和 Claude 对话,它会自动写代码并在服务器上执行
👉适合科研新手、非生信背景、第一次做 ChIP-seq 的朋友
服务器目录结构如下(准备好原始数据+参考基因组):
.├── 00.rawdata/# fastq.gz 原始数据└── ref/# genome.fa + genes.gtf
用聊天方式完成生信分析。你说一句,它做一步。你不需要懂代码,它全帮你写好
你只负需要和Claude对话
其余工作——包括命令、分析、判断、图表、结果解释——都交给它完成
第一步:告诉 Claude 你的目标❝我想做一套 ChIP-seq 分析,我是零基础小白,不会代码。 服务器当前目录下已经有原始数据和参考基因组:
00.rawdata/# fastq.gzref/# genome.fa, genes.gtf
请你从头开始一步步带我分析,每一步都: 1)用简单的话解释你在做什么 2)自动生成命令,先询问我后再执行 3)告诉我结果文件在哪里,是否正常 第一件事:请帮我创建分析环境(fastqc /fastp/ bowtie2 / bedtools/deeptools/macs3等)。
Claude 会自动安装所有软件,并告诉你环境准备完成。

继续把下面内容发给 Claude:
👇发送给 Claude:
❝下一步,请对 00.rawdata 下所有 fastq.gz 做 FastQC 质控,汇总生成 MultiQC 报告,结果放在新的目录。 做完后,请告诉我哪些指标正常、哪些可能需要注意,并用fastp做过滤。
Claude 会:
✔ 对每个样本跑 FastQC
✔ 汇总生成 MultiQC 报告
✔ fastp做过滤
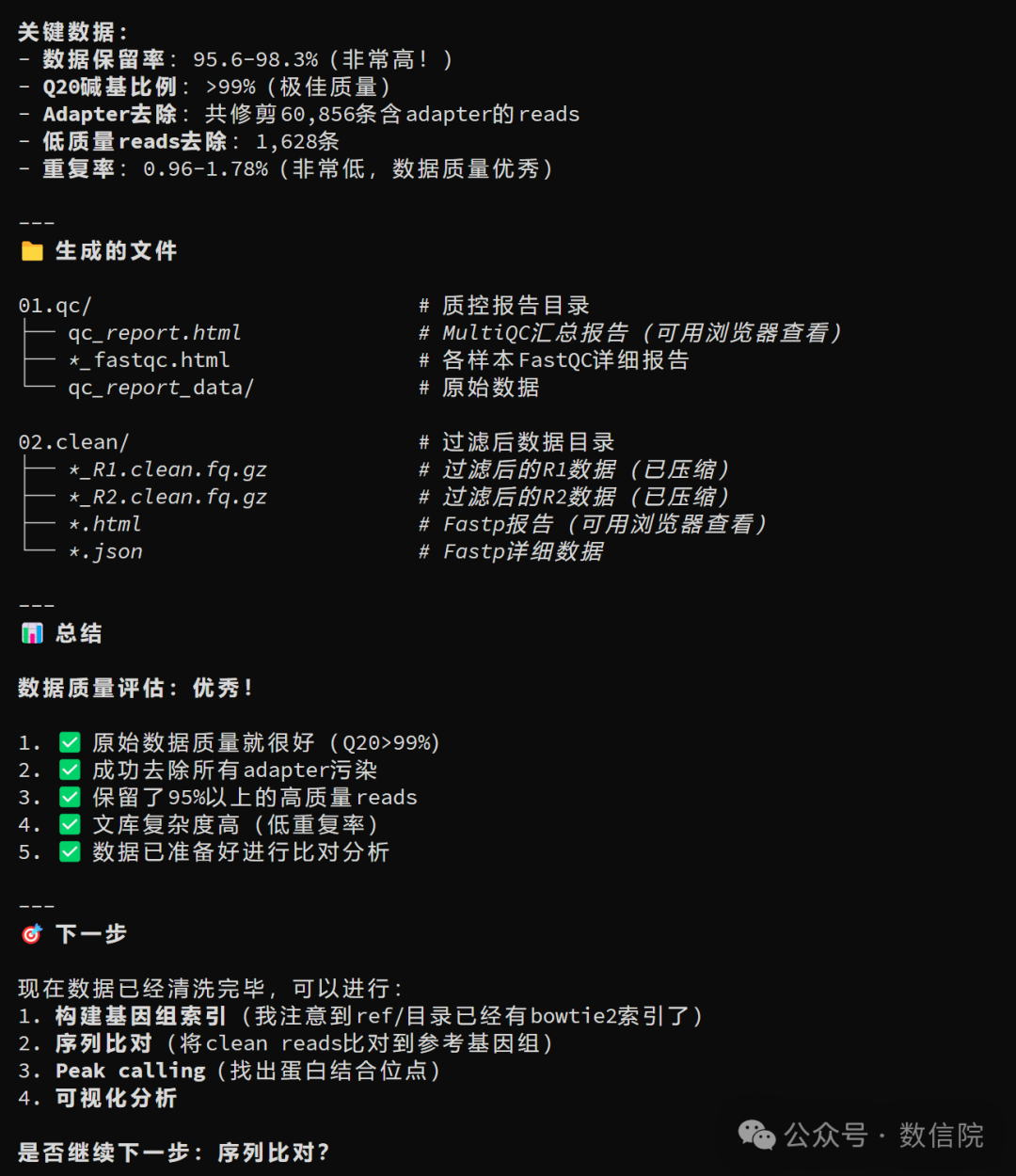
过滤后的数据和报告全部生成

继续发送:
❝下一步,请帮我把 ref/genome.fa 建立索引,并将所有 fastq数据比对到基因组。

比对完成

继续发:
👇发送给 Claude:
❝下一步用alignmentSieve对对比对后的数据进行过滤和去重--ignoreDuplicates --minMappingQuality 25 --samFlagExclude 260 --blackListFileName(blackList文件在ref目录下),结果放在新的目录。
开始运行

过滤完成

👇发送给 Claude:
❝下一步,将所有 过滤后的BAM 转成 bigWig,放在新的文件夹。
你会得到:
✔ 可视化轨道文件(bw)
✔ 适合上传 IGV 看的信号图

下一步,用deeptools做可视化分析,结果放在新的文件夹。
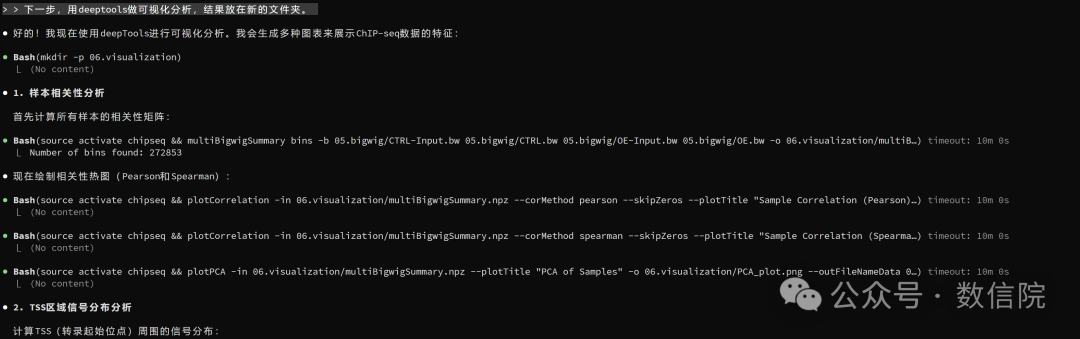
输出结果
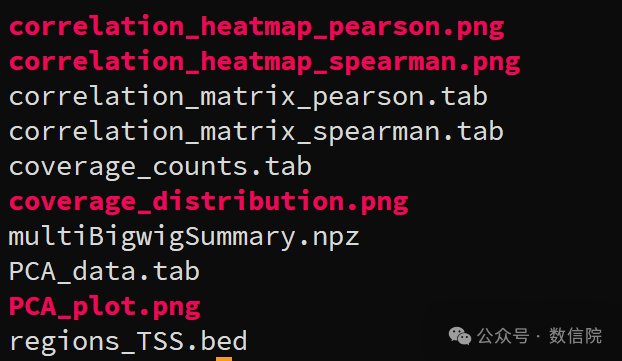
👇发送给 Claude:
❝用MACS3进行call peak,结果放到新的目录(自动处理input文件)。

继续发送:
👇发送给 Claude:
❝对peak做注释(分别使用HOMER做motif富集 和 ChIPseeker做注释), 结果放到新的目录。
Claude 会输出:
✔ 注释表格 ✔ 注释饼图 ✔ 启动子比例解释
分析结果目录:

👇发送给 Claude:
❝请帮我使用 DiffBind 做差异peak分析。 我不会 R,你写 R 代码并解释结果(火山图/聚类图也请自动生成)。
你将获得:
✔ DiffBind 结果 ✔ 差异峰表格 ✔ 可视化图

👇发送给 Claude:
❝请把完整分析流程自动整理成一份 Markdown 格式的 ChIP-seq 报告, 包括图、质控结果、峰数量、注释结果、差异分析、方法描述。 报告要适合发给导师和组会展示。
Claude 会给你一份:
✔ 专业 ✔ 完整 ✔ 图文结合 ✔ 可直接汇报的ChIP-seq 分析报告

claude真正实现了:
小白友好不需要会命令全程聊天式分析Claude 自动编写并执行命令自动诊断错误自动生成组会级别报告你只需把我给你的内容——逐步发给 Claude, 剩下所有操作它都会帮你完成。
🎉 写在最后随着Claude等大模型的不断进步,生物信息学领域将迎来更加智能和自动化的未来。大模型不仅为生信分析提供了无代码的解决方案,还推动了整个领域的智能化和高效化。科研人员将能够更专注于数据的深度挖掘与科学探索,而无需为复杂的技术细节担忧。
通过Claude,我们即将进入一个更加智能、高效的科研新时代。
🌱祝各位科研小白快速上手,轻松完成自己的数据分析!