晶格氧定义重要性、表征分析方法XPS、、XAFS、EPR、+(DEMS)晶格氧结构稳定性:电子关联性:DOI:10.1021/jacs.4c16801
二、晶格氧相关的核心公式与反应路径
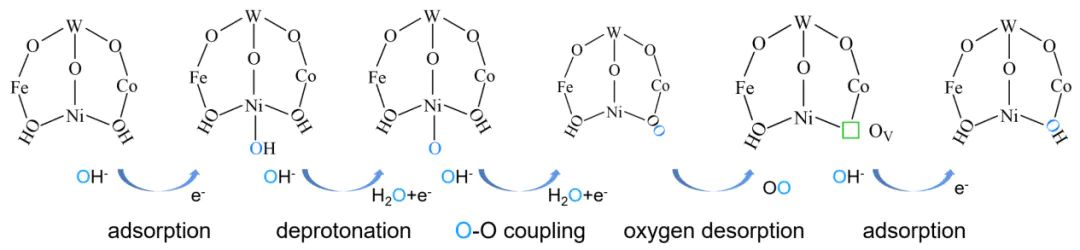
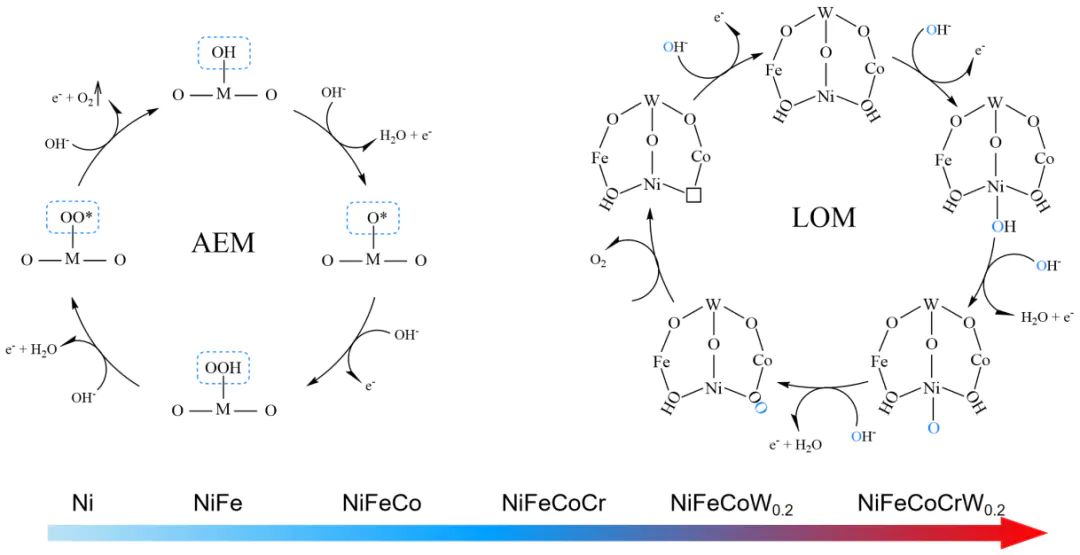
晶格氧参与的 OER 反应遵循多步电子转移与晶格氧释放耦合的路径,典型五步法反应机制如下:
电解液中的OH吸附至金属活性位点(M),完成首次电子转移
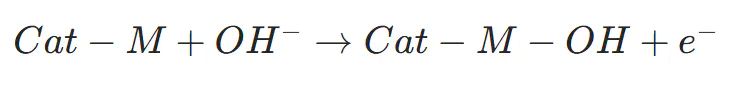
金属高价氧化步骤:(如Ni→Ni、Co→Co,n 为初始价态)
高价金属阳离子通过 d-p 轨道杂化打破相邻 M-O 键,释放晶格氧(O)并与表面M−O结合形成O−O键,同时产生氧空位(O)
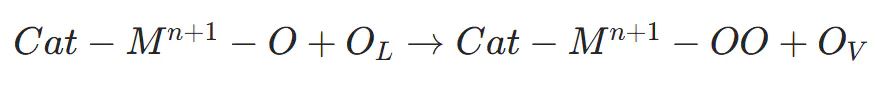
M−OO与电解液中OH反应,生成OOH−类中间体
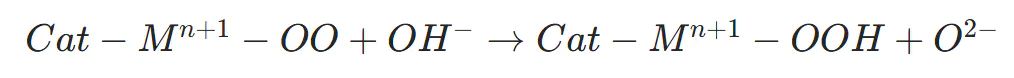
OOH−中间体脱氢释放O,金属阳离子还原至初始价态,完成催化循环
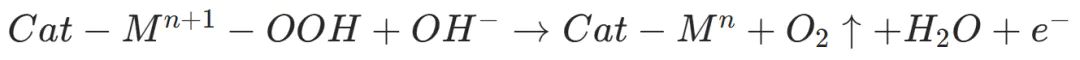
关键热力学与电子结构公式
公式简化为:
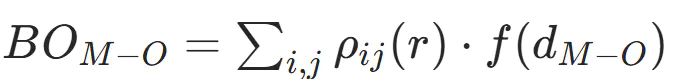
M−OM−ODOI:10.1038/s41467-025-58648-y
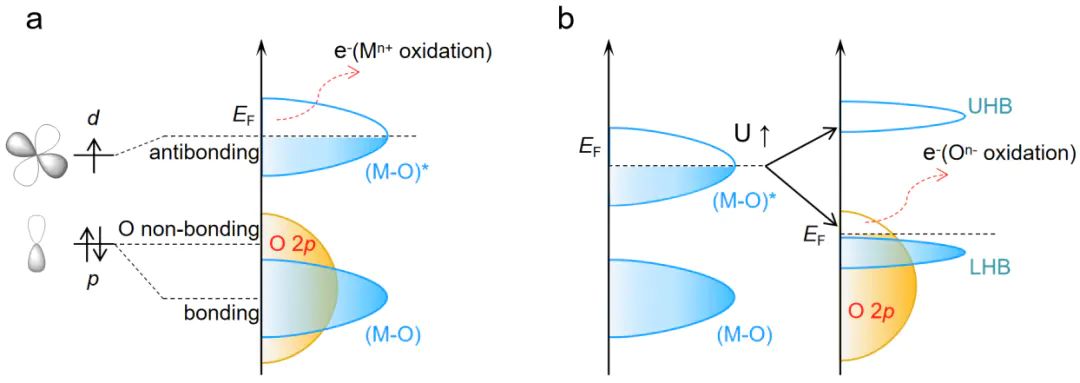
金属 d 轨道与氧 2p 轨道的能量差(ED)决定了电子转移效率,是晶格氧激活的关键电子结构参数,
其中,E为金属 d 带中心能量,E为氧 2p 带中心能量,两者均以 费米能级(E)为基准。ED 值越小,d 轨道与 p 轨道电子相互作用越强,越易发生电子转移,晶格氧活性越高。
Mott-Hubbard 分裂能(ΔELHB−p)
分裂能公式为:
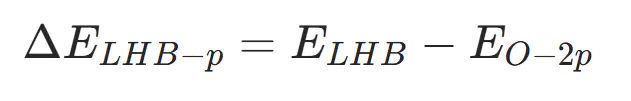
LHB−p
(EOV)
公式为:
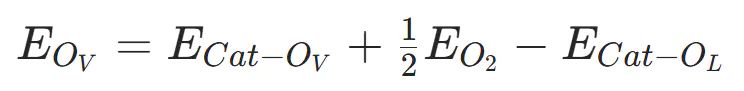
Cat−O为含氧量空位的催化剂总能量,E为氧气分子总能量,E为含晶格氧的原始催化剂总能量。E值越小,氧空位越易形成,晶格氧释放越容易。
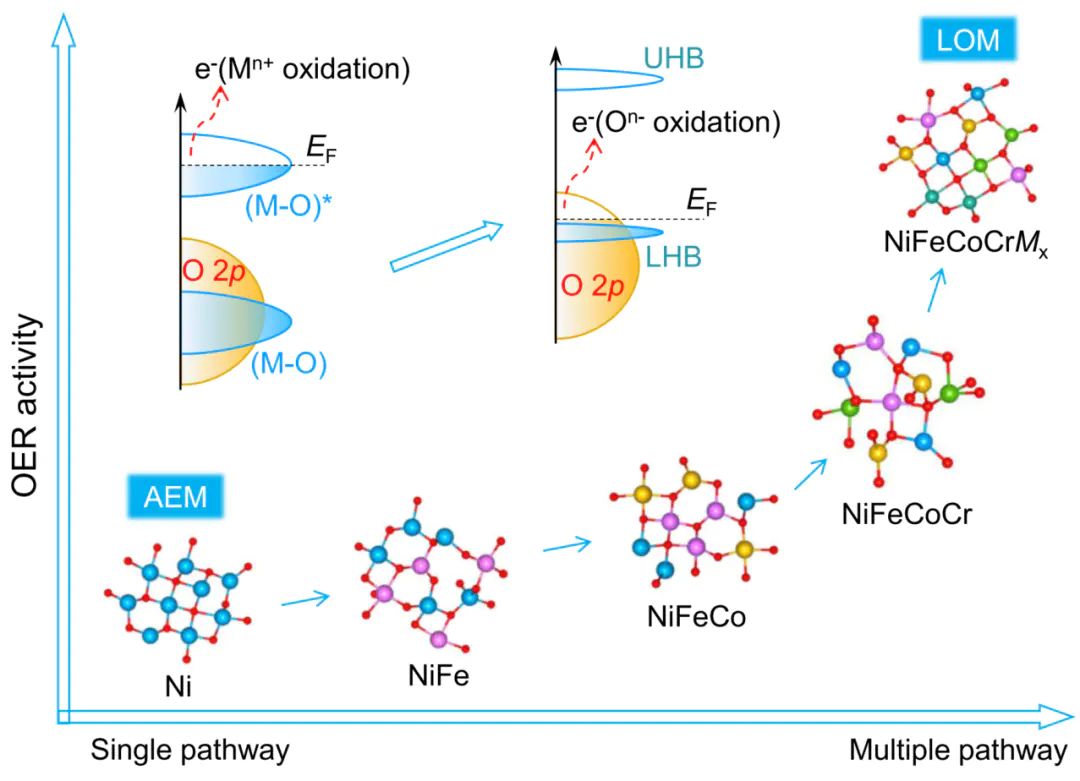
OER 反应是水分解、金属 – 空气电池等能源技术的速率控制步骤,其核心挑战是高过电位和慢动力学。晶格氧的激活通过以下三点解决这一痛点:
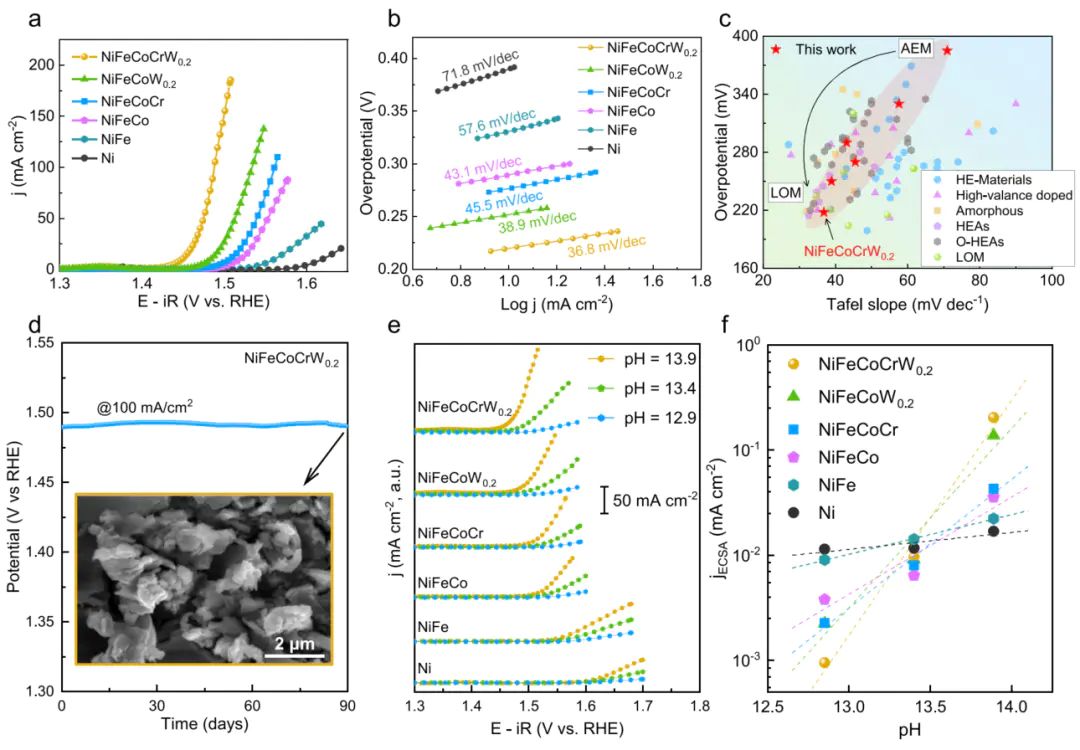
在 AEM 机制中,O∗与OOH∗的吸附能存在线性标度关系(scaling relation),无法同时优化两者吸附强度,导致过电位难以降低;而 LOM 机制中,晶格氧直接参与O−O键形成,绕开了这一限制,可将过电位降至 220 mV 以下。
2.提升反应动力学
DOI:10.1038/s41467-025-58648-y
传统 AEM 催化剂易因长期吸附中间体导致活性位点堵塞,或因金属离子溶出(如 Fe³⁺、Co²⁺)而失活;而 LOM 机制中,晶格氧释放产生的氧空位具有 “自修复” 能力,空位可快速吸附电解液中的OH,同时高熵合金的化学无序性抑制金属离子过度溶出。
要验证晶格氧的存在、活性及参与反应的证据,需结合与。
X 射线光电子能谱X 射线光电子能谱(XPS)羟基氧、吸附氧。
DOI:10.1038/s41467-025-58648-y
原位拉曼光谱,实时捕捉中间体生成与转化。在 LOM 机制研究中,关键特征峰包括:金属 – 氧键振动、O-O键振动。
DOI:10.1038/s41467-025-58648-y
¹⁸O 同位素标记差分电化学质谱(DEMS)22其中I、I、I分别为 m/z=32、34、36 的离子电流强度。
0.22DOI:10.1038/s41467-025-58648-y
X 射线吸收精细结构X 射线吸收精细结构(XAFS)XANES 通过吸收边能量反映金属价态(吸收边能量越高,价态越高);EXAFS 通过傅里叶变换(FT)得到径向分布函数(RDF),反映金属周围配位原子(如 O)的种类、配位数(CN)及键长()。
Ni K 边 XANES 显示,NiFeCoCrW的吸收边能量(8340 eV)比 NiO 标准样(8338 eV)高 2 eV,结合标准样校准,确定 Ni 的平均价态为 + 3.6(接近Ni),证明 W 的引入可显著提升 Ni 价态,增强 M-O 键的极化程度,利于晶格氧释放。
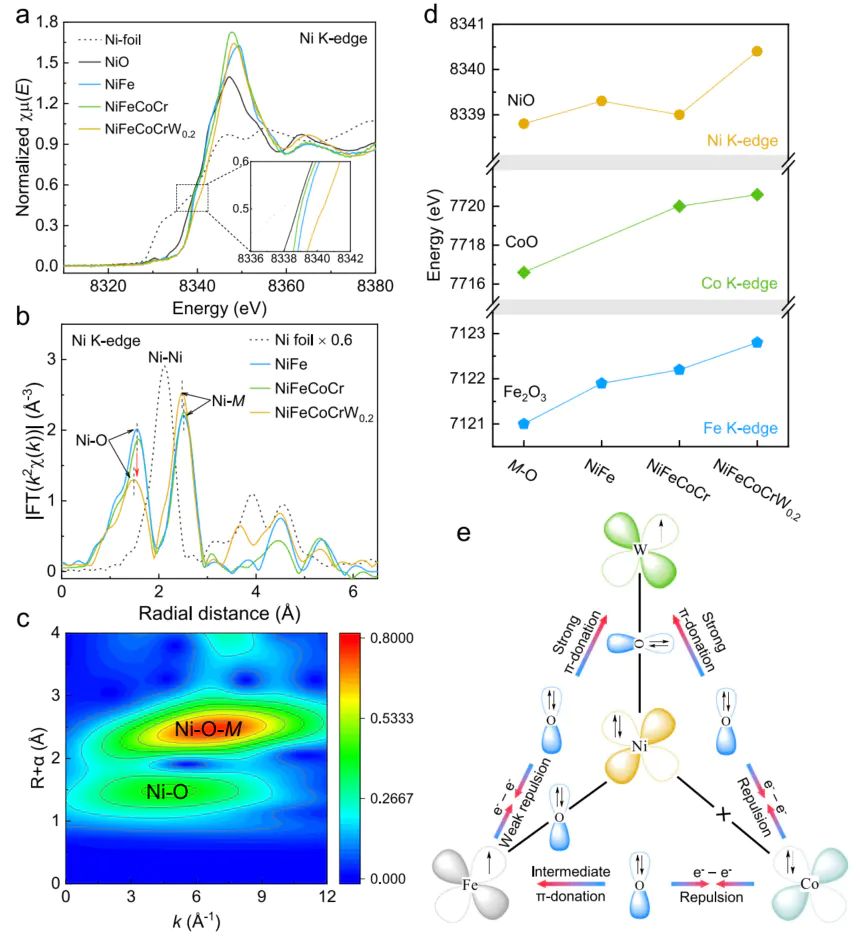
(EPR)
通过探测未成对电子的自旋信号(g 因子),直接表征材料中的缺陷(如氧空位O)。氧空位具有未成对电子,在 EPR 谱中表现为对称的洛伦兹峰,峰强度与氧空位浓度正相关。
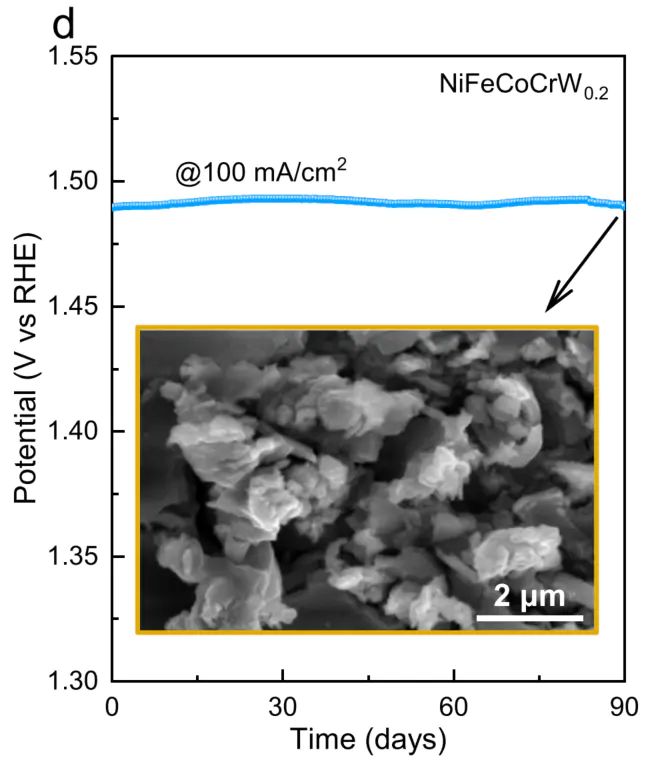
0.2DOI:10.1038/s41467-025-58648-y
电感耦合等离子体质谱电感耦合等离子体质谱(ICP-MS)金属离子过度溶出会导致催化剂结构坍塌,间接影响晶格氧的稳定释放;而适度溶出(如 Cr 的预溶出)可产生氧空位,促进晶格氧激活。
0.2DOI:10.1038/s41467-025-58648-y
DFT 计算
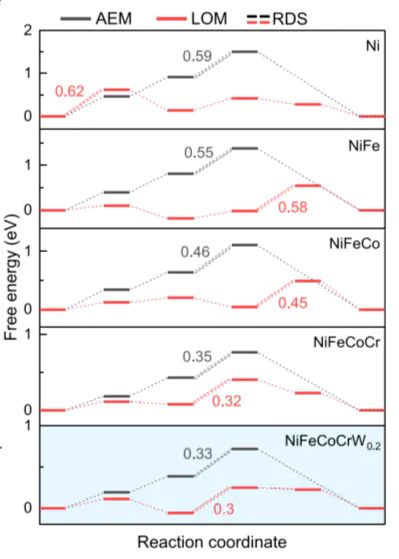
(DFT)自由能0.2DOI:10.1038/s41467-025-58648-y
对比 AEM 与 LOM 路径的。结果显示,NiFeCoCrW的 LOM 路径 RDS(O−O键形成)自由能(0.33 eV)低于 AEM 路径(0.55 eV),证明 LOM 为优势路径。
价金属离子(如 W⁶⁺)增强 d-d 库仑相互作用,使低哈伯德带(LHB)与 O 2p 带重叠,当ΔE论文中NiFeCoCrW的ΔE=−0.2eV,满足晶格氧激活条件。